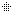هو اضطراب وراثي نادر، يتم وراثته عن طريق الوراثة السائِدة المرتبطة بX، ويصيب الجلد والشعر والأسنان والأظافر والجهاز العصبي المركزي. تمت تسميته بناءً على مظهره تحت المجهر.يتميز المرض بتشوهات الجلد التي تبدأ منذ الطفولة، وعادة ما تكون هذا التشوهات طفحًا متقرحًا قابل للشفاء، يليه نمو جلدي أصعب. قد يصاب الجلد ببقع رمادية أو بنية اللون تتلاشى مع مرور الوقت. يمكن أن تشمل الأعراض الأخرى تساقط الشعر، تشوهات الأسنان، تشوهات العين التي يمكن أن تؤدي إلى فقدان البصر والأظافر المخططة أو الوهداء.
يمكن أن تؤدي بعض المشكلات المرتبطة بالمرض التطور المتأخر، والإعاقة الذهنية، والنوبات وغيرها من المشكلات العصبية. معظم الذكور المصابين بالمرض لا ينجو من الولادة.
ينجم سلس الصباغ عن طفرة في جين IKBKG، الذي يشفر
بروتين NEMO، والذي يعمل على حماية الخلايا من الاستماتة المُحدثة عن TNF-alpha. وبالتالي فإن نقص الجين IKBKG يجعل الخلايا أكثر عرضة للاستماتة.
لم يتم اكتشاف علاج محدد للمرض؛ يجب أن تدار الظروف الفردية من قبل المتخصصين
تشكل مرض سلس الصباغ على طول خطوط بلاشكو
عند فتاة تبلغ من العمر 3 سنوات.
يمكن أن تحدث التشوهات الهيكلية والبنيوية في حوالي 14٪ من المرضى، بما في ذلك:
علم الجينات
يتم وراثة الIP بطريقة سائدة مرتبطة بX.
مرض سلس الصباغ قاتل في معظم الذكور وليس جميعهم. ربما تكون الأنثى المصابة بـ IP قد ورثت طفرة IKBKG من أي من الوالدين أو لديها طفرة جينية جديدة. قد يتأثر الآباء سريريًا أو لديهم فسيفساء جنسية (فسيفساء جنسية).
النساء المصابات لديهن خطر بنسبة 50٪ في نقل أليل IKBKG الطَفري عند الحمل؛ ومع ذلك، معظم الذكور تتسبب بالإجهاض. وبالتالي، فإن النسبة الفعالة للأطفال الذين يعيشون من الأم التي تحمل طفرة هي 33 ٪ من الإناث غير متأثرات، و 33 ٪ من الإناث المتأثرة، و 33 ٪ من الذكور غير المتضررين. تكون الاستشارة الوراثية واختبارات ما قبل الولادة والتشخيص الوراثي ما قبل الزرع متاحة.
في الإناث، تموت الخلايا التي تعبر عن جين IKBKG الطفري بسبب تعطيل الصبغي إكس بشكل انتقائي في وقت الولادة تقريبًا، لذلك فإن تعطيل الصبغي X- يكون منحرف للغاية. سبب IP هو طفرات في جين يسمى (NF-modB modulator)
التشخيص
يتم تشخيص IP من خلال النتائج السريرية وأحيانًا عن طريق خزعة الجلد. يكشف الاختبار الوراثي الجزيئي لجين NEMO IKBKG (موضع الكروموسوم Xq28) عن حدوث طفرات مسببة للأمراض في حوالي 80٪ من البروبينات. مثل هذا الاختبار متاح سريريا.
بالإضافة إلى ذلك، فإن الإناث المصابات بسلس الصباغ يكون لديهم انحراف تعطيل الكروموسوم إكس؛ يمكن لهذا الاختبار دعم التشخيص.
تعرض العديد من الأشخاص في الماضي لتشخيص خاطئ من النوع الثاني من IP، المعروف سابقًا باسم IP1. ولكن، الآن، أصبح هذا المرض يُعرف باسم آخر وهو سَلَسُ الصِّباغِ المُنْقِصُ التَّلَوُّن (Incontinentia pigmenti achromians).
هذا المرض له أعراض مختلفة قليلاً: دوامات أو شرائط من نقص التصبغ أو فقدان التصبغ، كما أنه لا يتم وراثة هذا المرض ولا يشمل المراحل الجلدية 1 أو 2. يعاني حوالي 33-50٪ من المرضى من تعدد الأنظمة - تشوهات العين والهيكل العظمي والعصبي. يكون موضع الكروموسومات في Xp11، بدلاً من Xq28.
العلاج
لا يوجد حتى الآن علاج محدد لسلس الصباغ. يمكن أن يُعالج العلاج الأعراض الفردية فقط.
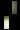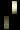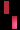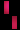





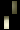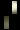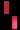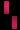
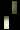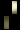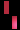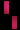









 عضويتي
عضويتي 
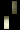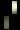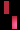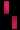





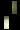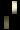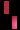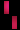
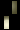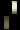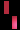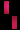
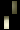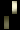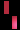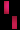
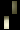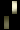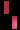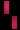




 العرض الشجري
العرض الشجري